文章来源:冠通检测一、医疗器械CE认证MDR指令是什么?MDR认证 2017年5月5日,欧盟医疗器械法规(REGULATION (EU) 2017/745,简称EU MDR)正式发布,并于2021年5月26号正式实施MDR中增加了很多新的概念,从MDD中的14个概念,增加到现在的71个,如增加了一些临床评价方面和上市后监管方面的概念法规规定:从2021年5月26号开始公告机构不能按照MDD颁发CE证书,目前I及以上风险等级产品认证机构已不再受理MDD指令的认证申请MDD失效时间:2024年5月27号 2024年5月27号起,企业持有的MDD指令的CE证书全部失效 请注意以下新更新通知:欧盟宣布推迟部分 I 类器械MDR合规时限 2017年5月,欧盟医疗器械新法规MDR (REGULATION EU 2017/745) 颁布,新的法规将替代原有的医疗器械指令 (MDD 93/42/EEC) 和有源植入性医疗器械指令 (AIMDD 90/385/EEC) 医疗器械CE认证(Conformité Européenne认证)是一种欧洲市场的合格性认证,用于确保医疗器械在欧洲经济区内销售和使用时符合欧洲法规和安全标准的要求CE认证是由欧洲联盟制定的法规和指令的一部分,适用于各种医疗器械,包括医疗设备、诊断设备、手术工具、体外诊断试剂和其他医疗器械对于目前获得CE证书的企业,应基于自身设备的临床证据的充分性合理安排申请MDR的时间,尽快启动MDR法规合规准备事宜 按照MDR法规要求厂家开始投入欧盟市场,产品进入欧盟需要做CE认证,满足欧盟医疗器械法规(MDR)要求医疗器械的相关法规文件:医疗器械指令是欧盟医疗器械的法规性文件目前,欧盟已颁布实施的医疗器械指令有三个,包括:1)有源植人医疗器械指令(AIMD, 90/385/EEC)该指令是针对任何可以通过内、外科方式,全部或部分植入人体,或者用医疗手段插入人体孔道,并旨在经此过程后留在人体内的有源医疗器械例如心脏起搏器、可植入的胰岛素泵、除颤器等该指令于1993年1月1日起生效,1995年1月1日强制实施要求自1990年6月20日开始认证,取得CE标志;在1994年12月31日以 后没有CE标志的有源植人医疗器械不得在欧盟市场上销售2)医疗器械指令(MDR, 93/42/EEC)该指令除有源植入医疗器械和体外诊器械外,几乎所有的医疗器械都属于该指令管理范围,如无源医疗器械(敷料、一次性使用产品、接触镜、血袋、导管等);以及有源医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等,于1995年1月1日起生效,1998年6月14日强制实施要求自 1993 年开始进行 CE认证,1998年6月13日以后没有CE标志的医疗器械产品不得在欧盟市场上销售2021年5月27日欧盟实施 Regulation (EU) 2017/745 on medical devices新法规,正式实施MDR 93/42/EEC Medical devices指令3)体外诊断医疗器械指令(IVDD, 98/79/EEC)该指令适用于血细胞计数器、妊娠检测装置等体外诊断医疗器械及附件于1998年12月7日生效,2003年12月7日强制实施该指令要求要求体外诊断试剂及仪器自1998年开始认证,取得CE标志2022年5月27日 Regulation (EU) 2017/746 on in vitro diagnostic medical devices体外诊断医疗器械法规正式实施,98/79/EC In vitro diagnostic medical devices IVDR指令开启欧盟市场新旅程拿到CE证书后,即可提交欧盟授权代表进行注册注册完成后就可以在产品上印CE标志和欧盟授权代表标志出口了
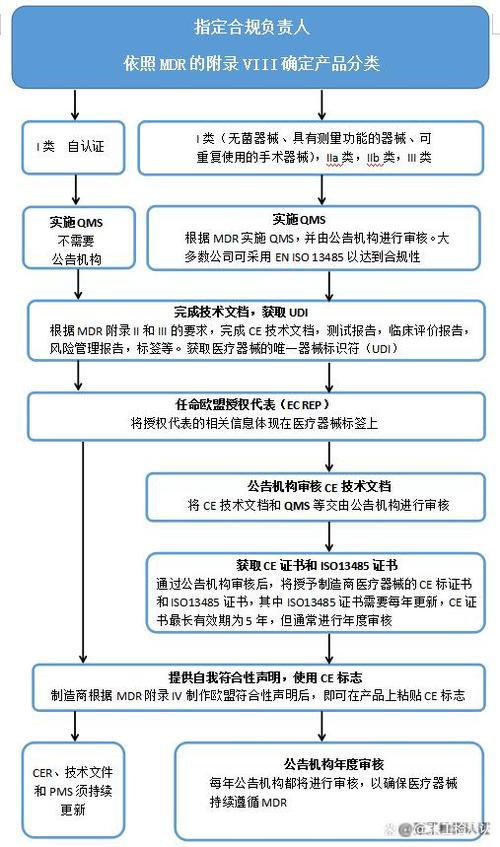
二、医疗器械CE认证的流程1. 确定医疗器械分类根据医疗器械指令的要求,医疗器械分为三类:I类、II类和III类不同类型的医疗器械有不同的认证要求和流程2. 准备技术文件技术文件是医疗器械CE认证的重要部分,包括产品说明书、技术规格、风险评估报告、用户手册等技术文件应详细描述产品的性能、使用方法和风险评估结果3. 提交技术文件技术文件应提交给欧盟认证机构进行审查认证机构是经过欧盟认可的第三方机构,负责评估技术文件和监督医疗器械的质量体系4. 认证机构审查机构将对技术文件进行审查,确认其是否符合医疗器械指令的要求如果发现任何问题,认证机构将通知申请人进行修改5. 符合性证书颁发如果技术文件通过审查,认证机构将颁发符合性证书符合性证书是证明医疗器械符合欧洲指令要求的证书,也是申请CE认证的重要依据三、MDR认证说明书的要求:一、说明书应同时满足以下要求:a. 符合医疗器械CE认证新MDR法规附录I中第3章的要求;b. 符合产品标准中有关说明书和标签的要求;c. 符合标准EN ISO 15223-1:2016, EN 1041:2008+A1:2013等相关标准要求四、医疗器械CE认证的影响市场准入:获得CE认证是产品进入欧洲市场的先决条件,有助于企业拓展欧洲市场竞争优势:拥有CE认证可提高企业在欧洲市场的竞争力,赢得客户的信任法规遵从:遵循欧盟相关法规和指令要求,降低违规风险,确保企业持续稳定发展品质保证:CE认证有助于提升企业产品的品质保证能力,提高客户满意度国际合作:CE认证在国际上被广泛认可,有助于企业开展国际合作,开拓更广阔的市场五、医疗器械CE认证常见问题在医疗器械CE认证过程中,企业可能会遇到以下常见问题:对标准的理解不足:企业需要对CE认证相关的标准进行深入理解,确保产品符合相关要求文件准备不充分:申请CE认证需要提交一系列文件,如产品技术资料、质量管理体系文件等,企业需要充分准备这些文件技术问题解释不清晰:在审核过程中,监管机构可能会对产品的技术问题进行询问,企业需要对这些问题进行清晰解释工厂审查不合格:企业的生产场所需要满足相关法规和标准要求,如果审查不合格,需要采取改进措施认证费用过高:CE认证费用较高,企业需要考虑如何在保证产品质量的同时降低认证成本医疗器械CE认证是进入欧盟市场必备的认证之一医疗器械CE认证的标准和要求是制造商必须遵循的一系列法规和技术标准通过严格的审核和测试,CE认证确保了医疗器械的安全性、可靠性和有效性,保障了欧洲人民的生命健康








0 评论