")
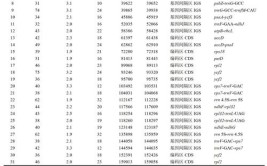
众所周知,小麦的基因组很大,很大,很大。这里的三个“很大”,分别指(1)普通小麦的基因组很大,(2)每个亚基因组也很大,(3)每个染色体也很大。相信大家都在IWGSCv1 参照基因组序列公开以后,就开始各种兴奋地进行BLAST,mapping,alignment。各位亲自玩过序列的同行,也一定记得,161010_Chinese_Spring_v1.0_pseudomolecules_parts.fasta这个文件(见我们在去年的推送:Ms1基因组重测序数据重分析--from sra to g.vcf.gz)(如下图)。这里有一个问题,大家可能注意到了:小麦基因释放的序列(_parts.fasta结尾的文件)中,每一条染色体是分成两部分的,比如chr1A_part1 和chr1A_part2.为什么要把每条染色体分成两部分?我们可以判断的是,每条序列两部分的分割并不代表短臂和长臂的分割点。如下表统计,我们可以看到,分成了两部分的各自长度都不超过500M。其实,根据大家的坊间经验,如果一个染色体的长度如果超过了500Mbp,那么很多已经通用的生物信息学分析工具是跑不通(或者至少很不顺利)的,如bowtie,bwa等。大家一定有过这个疑问:为什么超过500M就跑不通了?程序有什么bug吗?为什么很多基因组分析的程序都限制500M?其实小编也一直很疑惑。在此,小编用仅有计算机知识和大家一起探讨探讨可能性。我们先算一下log2(500M) 大约是多少?log2(500M)约为log2(512x1000x1000)约为log2(29x210x210)=29。估计这个阈值的上限应该不大可能正好是500M,而且很有可能是229=536870912,也就是约为536.9M。这个数字干嘛的?小编Google了下:无独有偶,小编发现很多关于这个数字的讨论,都和字符串的长度有关。看来,这个数字是很多程序在实现“字符串”(string)类型中的上限了。那么为啥是229呢?小编也不清楚,但可以用仅有的计算机知识帮大家猜一猜。毕竟,一个int(整数)类型在计算机的实现中,通常是4个字节的(4 byte)。而一个字节(1 byte)是8个比特(8 bit)(注意,这里不是比特币)。那么,用于编码一个int型整数的,通常有32个bit。考虑如果有3bit位被保留使用的情况,那么229正好就是上界限了。也可能和实现的程序语言中string类型的长度有关?也可能和后缀树的size上限有关?但不管怎么样,536870912似乎现在是很多程序在进行单个染色体序列(单个字符串)分析的上限了。在bowtie,Mummer和bwa等软件实现的时候,一定也没有考虑到会有物种的单个染色体超过这个536870912界限的情况。比如拟南芥、水稻、玉米、人的基因组..... 但单个染色体长度超过这个上限的,肯定包括:普通小麦、乌拉尔图小麦、硬粒小麦、粗山羊草、黑麦.....但是我们的小麦基因组研究已经进入了“春天”;但忽然就遇到了这样“一道坎”。难道我们需要修改程序的源代码去迈过这道“坎”吗?如果迈不过去,那我们先绕过去吧~如果后面还有什么坑、什么坎等着我们:不用怕,咱们小麦人一起同在




0 评论