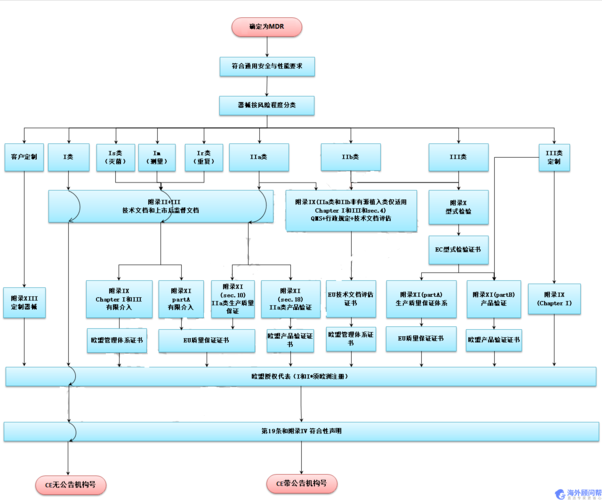
MDR法规简介:MDR (REGULATION EU 2017/745)是欧盟关于人类用医疗器械临床研究和销售的法规,该法规的目的是确保更好地保护公众健康和患者安全依据MDR Article 123的要求,MDR于2017年5月26日正式生效,并与2020年5月26日期正式取代MDD(93/42/EEC)和AIMDD(90/385/EEC)自申请之日起,所有在欧盟市场上新投放市场的医疗设备都必须符合MDR的要求MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性依据Article 120 clause2的规定,过渡期内NB签发的CE证书继续有效,但是从其交付日期起有效期不超过5年,并且于2024年5月27日失效MDR的主要变化:1.扩大了应用范围2.提出了新的概念和器械的定义3.细化了医疗器械的分类4.完善了器械的通用安全和性能要求5.加强对技术文件的要求6.加强器械上市后的监管7.完善临床评价相关要求8.提出Eudamed数据库的建立和使用9.提出器械的可追溯性(UDI)10.对NB提出严格的要求新法规对医疗产品进口商有哪些变化?1.进口商在将制成的医疗设备投放到欧盟市场之前,必须对制成的医疗设备执行标签控制(标签和使用说明),以确保所有设备均配备了UDI向量(条形码或矩阵代码)2.进口商应在欧洲EUDAMED数据库中将设施注册为“进口商”3.如果设备存在实际或潜在问题,则进口商必须通知制造商,授权代表以及必要时通知主管当局分销商有哪些变化?1.分销商保证设备的存储和运输条件适当并符合制造商的建议2.最后,分销商会注册索赔,不合格的设备并帮助召回进口商和分销商必须实施并保持最新的质量管理体系,以履行《 MDR》第13、14和25条所述的义务MDR医疗器械产品类别:所有器械通过医疗器械指令(MDR)附录IX的分类规则被划分为四个管理类别:I类,IIa类,IIb类和III类I 类医疗器械:通常是指不接触人体或只接触完整皮肤的医疗器械I-m(测量)医疗器械:带有测量功能的I 类医疗器械I-s(灭菌)医疗器械:最终以灭菌形式出现在市场上的I 类医疗器械I-r(重复使用外科)医疗器械:不连接到任何有源医疗器械,制造商预期可通过适当的处理之后再次使用IIa 类医疗器械:风险等级较一类医疗器械高,一般是指暂时使用的侵入器械等,有能量交换或测量的有源医疗器械IIb 类医疗器械:风险等级较高,一般指会对人体有潜在风险或者是长时间使用III 类医疗器械:风险等级最高,一般用于人体中枢循环系统或大脑如果软件用于为诊断或治疗提供信息,属于IIa类除非此类决定有以下影响:患者的死亡或不可逆转的健康恶化,属于III类;健康状况或外科手术干预下严重恶化,属于IIb类;用于监测生理过程的软件属于IIa类,除非用于监测重要的生理参数,并且这些参数的变化可能对患者造成直接危险,这种情况下,它是归类为IIb类;其他种类的软件将被划为I类MDR认证流程:1.项目申请——递交CE认证申请表2.资料准备——根据CE认证要求,企业准备好相关的认证文件3.产品测试——企业将待测样品寄到实验室进行测试4.编制报告——认证工程师根据合格的检测数据,编写报告5.递交审核——工程师将完整的报告进行审核6.签发证书——报告审核无误后,颁发CE认证证书MDR认证办理:沃证(VIACERT)国际检测认证中心是欧盟公告机构的大中华区办事处,能为您详细解读医疗器械MDR新法规,提供培训、辅导、认证一站式技术服务,已为众多企业申请医疗器械CE认证





0 评论